Phenylketonuria is a non-delaile disease that can lead to dementia. However, this ailment is treated. In some cases, only the correct diet is sufficient, and the child with phenylketonuria will develop normally. To do this, you need to recognize the disease. Learn how this ailment is diagnosed.
Content
What is phenylketonuria

Analyzes. Among them - blood test on phenylketonuria. Despite,
that medicine is developing rapidly, humanity has not yet learned to
to fully prevent these hereditary pathologies and completely
heal, so much depends on the timely assigned
Diagnosis.
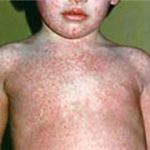
Externally, such children, as a rule, blond with blue eyes, very sensitive light skin, they often show eczema, dermatitis, various rashes. The physique is disproportionate, the vices of the development of internal organs are often detected - heart, liver, etc. From the point of view of biochemistry, phenylalanine is an indispensable amino acid, a kind «Brick», The necessary human body for constructing a protein molecule. With phenylketonuria, the formation of the most important biologically active substances necessary for the normal operation of the brain, especially serotonin and dopamine, is reduced. They are launched «breakdown» In the mechanism of mental development of a child leading to the progression of dementia.
Risk group
Symptoms of phenylketonuria
External signs of phenylketonurium can sometimes be suspected in the first weeks and months of the life of the baby, although they are very different, and sometimes such babies can even differ from healthy: they, for example, are also well added in weight. And others, on the contrary, from the first days after the birth of sluggish, Sonline or Restless, do not fix a look at the items, do not turn to the sound source, it is late to grow and do it inexpressively.
An important and characteristic early symptoms of the disease include a specific unpleasant smell of mold or mouse, emanating from urine and child skin. Such children are often poorly absorbed by writing, they have abundant jumping, later, in the second half of their life, they are lagging behind in psychomotor development: the development of motor skills, speech and t.D. Mental Delay Delay is the main sign of the disease.
Diagnosis of phenylketonuria
In the case of suspected illness, the answer comes to a children's clinic in which a child is observed. Also to identify in the urine of phenylpirogradic acid and other ketone acids, appearing by the end of the 1st week of the life of the baby, the indicator papers are used «Phoenistix», «Biophan». They enter the mandatory set for express diagnostics of each precinct pediatrician. The test is carried out repeatedly during the 1st year of the life of the crumbs in those families where children born with phenylketonuria. So, over the age of 4-6 weeks in the urine of a sick child can easily reveal the products of the decay of phenylalanine.
These and other similar methods refer to the categories of approximate, therefore, with positive results, a special examination is required using accurate quantitative methods for determining the content of phenylalanine in the blood and urine, which are carried out by centralized biochemical laboratories. Children who are not included in the risk group (in families where there were no cases of phenylketonurium), blood test is carried out once in the maternity hospital.