Phenylketonuria is a congenital disease. In the majority of untrenented patients, the disease is manifested by one degree of mental retardation. What is phenylketonuria? Read about it in the article.
Content
 Phenylalanine refers to indispensable amino acids (substances that go to the construction of proteins in the human body), since animal fabrics are not capable of synthesize. In the human body, where phenylalanine falls with food, the process of its oxidation is under way using the specific enzyme phenylalannine hydroxylase to a completely replaceable amino acid - Tyrosine. Violation (blocking) of this reaction, observed in violation of the synthesis of phenylalananinhydroxylase in the liver, leads to the development of severe hereditary disease - phenylketonuria.
Phenylalanine refers to indispensable amino acids (substances that go to the construction of proteins in the human body), since animal fabrics are not capable of synthesize. In the human body, where phenylalanine falls with food, the process of its oxidation is under way using the specific enzyme phenylalannine hydroxylase to a completely replaceable amino acid - Tyrosine. Violation (blocking) of this reaction, observed in violation of the synthesis of phenylalananinhydroxylase in the liver, leads to the development of severe hereditary disease - phenylketonuria.
Tyrosine is used by the body for the synthesis of thyroid hormones. And melanin, tyrosine and phenylalanine derivative, provides pigmentation (color color) of the skin, eyes and hair.
Phenylketonuria is also called phenylalaninemia, phenylpyrogradna oligophrenia. The disease relates to congenital metabolic disorders (metabolism), is characterized by an increase in the level of phenylalanine in blood plasma and is accompanied by mental retardation. The exact cause of mental retardation in phenylketonuria is unknown, but it is a consequence of a biochemical defect. The hereditary nature of the disease is found in most people. The frequency of occurrence of FCU is 1 case by 6000-10000 newborns.
Symptoms of phenylketonuria
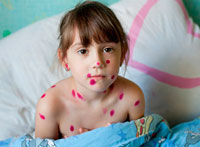
Clinical symptoms of phenylketonuria in newborns are usually absent, so laboratory screening should be carried out (diagnostics). In rare cases, lethargy is noted and difficulty. In the majority of untreneable patients, the disease is manifested by one degree of mental retardation (more often pronounced), which is the main symptom of phenylketonuria. Usually patients with brighter skin, hair and eyes than their healthy relatives. Some children may have skin changes that resemble infant ecase.
For patients with phenylketonuria, abundant neurological symptoms are characterized, reflexes are especially changed. In older children, both large and small epileptic seizures are noted, deviations on the electroencephalogram are noted in 75 - 90% of cases. A pronounced increase in activity and psychosis develops, often from patients comes unpleasant "mouse" The smell caused by the presence of phenyloxic acid in the urine and sweat.
Diagnosis of phenylketonuria
After the newborn received milk (the source of phenylalanine) for at least 48 hours, the doctors conduct a screening test on phenylketonuria (examine blood drop). At the age of 4 - 6 weeks, the product of the decay of phenylalanine can be revealed in the urine of the patient. At this time, one more screening test on phenylketonuria is carried out. The blade is carried out after the end of the newborn period, and children from families where there is a patient phenylketonuria, it should be regularly repeated during the first year of life.
Treatment of phenylketonuria
The treatment is to limit the admission of phenylalanine with food in order to meet the need for this indispensable amino acid, but not more; Ensure normal growth and development, not allowing accumulation in the body of phenylalanine and the final products of its exchange. Permanent monitoring is required for the level of phenylalanine in the child's blood. In the United States to replace milk, the product Lofenalk is widely used, full in all respects containing a small amount of phenylalanine. It is allowed to use low-facular natural products, such as fruits, vegetables, some cereals and t.D. The need for phenylalanine is satisfied due to the consumption of a limited number of natural protein and the residual amount of phenylalanine in Lofenal. Currently, there are products that are fully purified from phenylalanine. The Phenyl Fri product contains all the necessary ingredients except phenylalanine, does not contain fat, therefore its energy value is lower than that of other products.
Physician treatment should be held from the first days of life. Only early and consistent treatment prevents the defeat of the central nervous system and severe mental retardation. If the treatment has begun after 2-3 years of life, it can only be limited to pronounced increased activity and facilitate the course of convulsive syndrome.